Please wait...
Good morning, everyone! I’d like to welcome you all to SPEX CertiPrep’s Webinar, “Identifying and Quantifying the Uncertainty Associated with Classical Method (Titration)”. My name is Amy Williams and I am the marketing manager of SPEX CertiPrep and I’ll be moderating the presentation today.
This is part two of a series on Uncertainty. Last year we presented a webinar on the Calculation of Uncertainty by an instrumental method such as ICP and this webinar is already available on our YouTube channel. Since then, we have had many requests for presentations to include other methods and we are pleased to present that to you today.
Before we begin, I’d like to give a few housekeeping items out of the way. Everyone in the attendance today will receive an email with the presentation slides and links to the webinar recording on our YouTube channel. If you have any questions during the presentation, please type them in the question box on your screen and we’ll answer as many of them as possible during our Q&A session. If we don’t answer your question, we will contact you after the webinar with an answer.
Now I am glad to introduce Vanaja Sivakumar. Vanaja received her PhD from the Department of Inorganic and Physical Chemistry from the Indian Institute of Science in Bangalore, India. She worked with Weaver Brothers India as a research chemist for 9 years before coming to SPEX CertiPrep 20 years ago as the QA and Regulatory manager. She served as SPEX CertiPrep’s Vice President of Inorganic Manufacturing and is now a Quality Management System Auditor with underwriters laboratory.
Vanaja:
Thank you, Amy. Good morning, everyone. As Amy mentioned in her introduction, this is the extension of the series on Uncertainty. In the first lecture, we talked about calculation of uncertainty from instrument method. There we explained various statistical parameters that would be used in calculating uncertainty. I would like to start with the same concept for this presentation as well since there could be some viewers watching this for the first time.
In the past, chemists focused on precisional process from any given method. However, this day’s not just enough to have a precise method. We have to establish the quality or accuracy of results by stating the measure of confidence.One useful measure for this is measurement uncertainty since precision, accuracy, confidence limit, or interval, errors in measurement are fundamental to analytical chemistry. We will be covering them here when we briefly. Main goal or test of this presentation is to explain about uncertainty in practical terms. We’ll be showing one of the ways to calculate uncertainty. Taking example from standardization of sulfuric acid with sodium carbonate. We will calculate uncertainty associated with the measurement as applied to the process we have chosen. True value for the quantity measured depends on method, instrument or measuring device, nature of measurements, and skill of the operator. Any of these could introduce errors in measurement. You can be confident about the results from validating method and a well-maintained instrument, a well-trained operator applying his or her mind to the task at hand and through experience can gauge the quality of the results. In spite of all these, an experienced chemist, precise instrument, and a validated method, it is hard to get the true value of measurements accurately. There are always errors and uncertainty associated with the measurements. So what are these errors?
Typically, there are three types of errors: Determinate Errors, Indeterminate Errors, Spurious Errors. Determinate Errors can be avoided if they are recognized. Unpredictable radiations in repeated observations of the measured value is one example. Errors associated with this are consistently bigger or smaller. This can be reduced by increasing the number of observations. Indeterminate errors or random errors, on the other hand, exist by the very nature of measurement data. The analyst does not know of their magnitude nor can he control it. These indeterminate errors affect the precision and accuracy of all chemical work. One such example is error due to inadequate control of experimental conditions. This can give rise to systematic errors that are not constant. These errors can be eliminated totally. However, we should attempt to contain them as narrow as possible. Other possible errors are Spurious Errors, these are due to human or instrument failures. Whatever be the type of errors, we have to eliminate or minimize them. One way to minimize the Determinate Errors is to make a series of measurements on the same object and record the average.
What is an average? When we do an experiment, we take a number of times measurements and calculate the average value from all these replications. It is the sum of all the individual measurements divided by the number of measurements. As we all know, average represents the central tendency wherein lies the true value. It’s not an absolute value. It’s only an estimate of true value. So how can you estimate the true value? One way to do, though it’s approximate, is through standard deviation represented sigma or S and you express true value with standard deviation.
So what is standard deviation? This is represented by this formula where X is the measurement data from that individual measurement; x-bar is the average of the volume measurement that you took and n is the number of replicates. Standard deviation shows how much variation or dispersion exist from the average or expected value. I said earlier, an average gives you estimate of true value. This is called point average. By using standard deviation, you can pinpoint the range within which this true value lies at a certain confidence interval. The term “confidence interval”, “confidence level”, “confidence limits”, and “interval estimates” are used interchangeably. So you express the true values as average x-bar +/- 1.645 at 90% confidence interval or x-bar +/- 1.96 at 95% confidence interval or x-bar +/- 3.091 at 99.8% confidence interval.
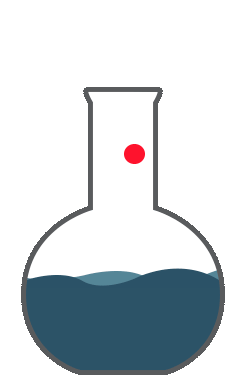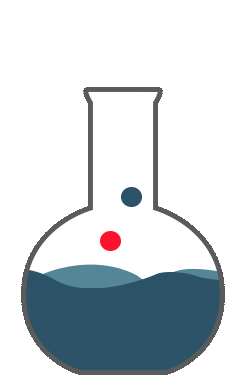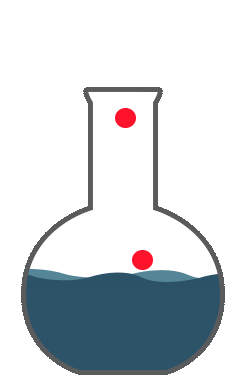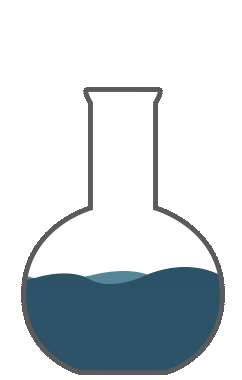
So you are expressing the true value by the dispersion of the standard deviation; now that represents your accuracy.Accuracy is the correctness of measurement in relation to true value. Let us examine these charts. Say, you want to do a titration and your endpoint of the titration that responds to 8.5ml to give an accurate equal point. You perform 8 replicates. The red line here represents the true value. The green or the blue indicates the values obtained by measurements. The first graph, you see the average of 8.55 which is close to the true value of 8.5. However, there is a dispersion around the true value. This is a good accuracy but precision is not so good.
The second graph, you would get an average of 7.39. The measurements are far below the true value. There is a consistent error giving a bias towards the low value. Hence, this is not accurate. So you're good with precision which is the reproducibility of the method. The good graph shows you the intended value of 8.5 which is close to the 8.55 and the replicates showed closeness to true value. This is good accuracy and good precision. The fourth graph you got here, you got the true value almost close to the true value 8.55, by way of the averaging number of measurements. However, the values are all scattered all over the place. You can’t have confidence in these measurements. So this scatter is represented by variance, standard deviation, or average deviation. Average deviation is a measure of dispersion or scatter around the true value and is given by this formula. The smaller the average deviation, the more precise is the measurement. However, it’s not an accurate measure of precision. Variance and standard deviations are better tools for indicating the spread of measurements around the true value. Variation of all these equations are given in any standard statistics. So all the parameters like standard deviation or multiple sub-standard deviations such as 2s or 3s or width of the confidence interval show the dispersion of the cells around the true value. This dispersion is the uncertainty around the true value or the measured value. Uncertainty is a parameter associated with the results of measurement. This characterizes the dispersion of values that could reasonably attributed to the measured value.
In general, the word uncertainty relates to gentle concept of doubt. The doubt is certainly not about the validity of the measurement. In fact, knowledge of uncertainty increases the confidence in the validity of the measurement result. At this point, it’s important to distinguish between error and uncertainty. Error is a single value and cannot be known exactly. Whereas, we can estimate the uncertainty.
Uncertainty estimation is simple in principle. We can follow your 5-step approach to calculate uncertainty. As a first step, we should have a clear idea of what is to be measured. We should review all the errors that could be corrected and eliminate them. Write down all the processes, use the reference material. What is the sample dilution? What are the devices used? What are the environmental conditions? Is there any uncertainty associated with the equipment? Gather all these information. Now, identify and quantify uncertainty from each component in this process.
Information given in the step 4 will consist of number of quantified contributions to overall uncertainty whether associated with the individual socials or with the combined effects of several sources. The contribution expressed as standard deviation should be combined according to appropriate rules to give combined standard uncertainty. Then, it should be expanded by applying your coverage factor. We will go in details for each of this step.
For quantifying uncertainty as a first step, you should know what type of uncertainty it is. Is it Type A or Type B? Type A is associated with repeated measurements usually like replicates in titration or average in many readings in instruments. Type A uncertainty is expressed by this formula here. Type B on the other hand, is based on scientific judgment using all the relevant information available including previous measurement data, manufacturer's’ specifications, or data provided in the calibration report. There are three common models for Type B; they are rectangular, triangular, and normal. Each one has a different normalizing factor. To calculate this standard uncertainty from the listed uncertainty, we apply this normalizing factor. We will review these factors for each model.
Rectangular distribution is used when uncertainty is stated without specifying a level of confidence. When there is no confidence stated, there is no reason to expect extreme values. Normalization factor is 1 divided by square-root of 3. So to convert to standard uncertainty, you would divide the listed uncertainty that you know by the square-root of 3.
The second model is triangular. Here, the distribution is symmetric and measured value lies close to the target value. We use this distribution when we take into account uncertainty associated with volumetric glassware. For example, you have a class A volumetric glass of capacity 500mL, listed tolerance from the manufacturer for this is +/- 0.2 mL. The normalizing factor would be listed uncertainty divided by square-root of 6. This is what you would use to convert the tolerance to a standard uncertainty. This model is based on normal distribution. This is used when we make an estimate from repeated observations of randomly varying process and express the results in certain confidence interval. Example, if you certificate sometimes that you have the uncertainty stated with the stated level of confidence, they might give you a coverage factor that is “k” used for coverage factor. So they may say this coverage factor is 2 or 2.08, if not, it’s not given and just stated at 95%, you might want to use 2s or 3s.
So far, we reviewed the types and models we used for uncertainty associated with each task. Now, we combine all of them based on the following principle. To combine uncertainty from each component for a particular source, we use model number 1. For example, if you have a pipette, you will use the uncertainty found the volume from the manufacturer as well as the temperature so you will be combining the components of uncertainty for pipette by this model. These interim uncertainties could be combined to give an overall uncertainty and this is the model that we use. Here we use what you call the relative standard uncertainty. It is observing chemical measurements dominant contributions to the overall uncertainty vary in proportion to the level of analytes. So it’s sensible to use relative standard uncertainty when they combine from each source. Although, the combined uncertainties used to express the uncertainty of many measurement results, it is required to define an interval above the measurement results. The measure of uncertainty in that intended to meet this requirement is termed the expanded uncertainty and this is done by the coverage factor k. So expanded uncertainty equals kuc. Coverage factor depends on confidence interval and number of observations. For most purposes, it is recommended that k is set to 2 for observations of greater than 7. For lesser observations you could use two-tailed values from student’s t. Once you expand the combined uncertainty, you would express the true value as average +/- combined expanded uncertainty.
So these are the terms we will be using to express uncertainty from the measurements that we are going to show you now: Average, Standard Deviation, Combined Uncertainty, and Expanded Uncertainty. So far we have covered the theory behind the uncertainty calculation. So let us use a practical example, for instance, a titration process. And see how we can proceed to quantify uncertainty associated with this. So we will base this on the determination of normality of sulfuric acid with sodium carbonate.
So let us review the steps. What are the steps would we follow. We would follow the five step approach that I discussed earlier. So what? In this case, we want to determine the strength or the normality of the sulfuric acid we prepared. What is the process? It’s a simple acid-base titration. A known concentration of acid is titrated against the known concentration of alkali to arrive at an equal end point. What are the sources of uncertainty? The uncertainty associated with the preparation of a standard, the uncertainty associated with the titration, repeatability and so on will be the sources of uncertainty. We will be going in detail in the next few slides. Once identified we’ll both calculate uncertainty for each of these processes. Finally, we will combine them from each of these steps based on the principle we stated earlier.
Normality of the sulfuric acid that we both determined is.. you arrive by this with the use of this equation: N2 equals V1 N1 divided by V2. By plugging the values we got in our experiment, we arrived at the value +/- 0.02499N. Here, the value for V1 is the volume of sodium carbonate. N1 is the normality of sodium carbonate. V2 is the volume of sulfuric acid that we already got and N2 is the normality of sulfuric acid. Now, these values that we have, finally as an average, also is associated with the uncertainty from all these processes. So what is the process, what is the symbol that we are going to use to represent the process? For the volume of sodium carbonate that we will use, we want to use the symbol uV1. For the uncertainty associated with the preparation and determination of normality of the sodium carbonate, we want to use the symbol uN1. For the sulfuric acid volume, we want to use the symbol uV2. The normality that we arrived at by way of average is the combination of all these uncertainties and that we represent by uN2.
So, I would like to present here the fishbowl diagram or a cause and effect diagram. The aim here is to identify all the major uncertainty sources and to understand their effect on the analyte and its uncertainty. As I mentioned earlier, we have four components here, uN1 coming from sodium carbonate, uV1 coming from sodium carbonate, uV2 coming from sulfuric acid, and u repeatability from the overall process, the combination of all these is leading to the uncertainty of the normality that we determined.
So what are the components for the uN1? You take certain amount of sodium carbonate. You make it up to volume so there is a weight component associated with it. There is a volume component here. The volume itself has 2 components: tolerance and temperature. Then with molar mass of sodium carbonate, that will give you some uncertainty and the certificate that we used, the compound that k will precipitate as impurity so that impurity from the certificate so uN1 is a combination of all these interim uncertainties. uV1 is the, we use a pipette to deliver a volume that has two components. One is the temperature coefficient of the volume expansion for glass and the tolerance associated with the pipette. So uV1 is the combination of these 2 interim uncertainties. Now uV2 sulfuric acid is again very similar to uV1 when we use a burette here instead of a pipette. Again it has a component from tolerance of the burette and the temperature coefficient. Then overall, we repeat this process many times to arrive at the average so that gives you the uncertainty and we will add all these together to arrive at this.
So let us examine how to calculate uN1. We use NIST SRM 351a for preparing standard solution. We weighed, we dry the material for the number of hour stated in the certificate, and we weighed exactly 1.0386 g in a balance and then we diluted it into 1 liter in a volumetric flask. There are 3 uncertainties associated with this process. What are they? Weighing on the balance, making up to volume in a volumetric flask and the impurities that we got from the certificate, the SRM certification.
So let us consider the first one, weighing on the balance. The uncertainty for the balance calibration was the listed uncertainty divided by the normalizing factor for rectangular distribution. We use rectangular distribution here because the balance certification that we had did not have this stated confidence interval. If you have the balance calibration certificate, with this stated level of confidence or a coverage factor, we could use the rectangular distribution. So you have arrived at the standard uncertainty for the balance but the standard uncertainty, the weighing process itself, has two process. It is done in two stages... Hence, uncertainty in the balance is a combination of these two processes. So you calculate the standard uncertainty by combining these two. Once you have the standard uncertainty, you divide it by the value that is this - this is the amount that we weigh. So this table here gives you in a nutshell all these formulas and values. So what is it, the device was balanced, the value is 1.0386g that we weighed, the standard uncertainty that we got which is similar to the, which is this. There is no other component to be added to this so the combined uncertainty is the same as the standard uncertainty. This will be compared to you when I talk about the uncertainty from the pipette. So currently, for the balance, we have the combined uncertainty same as the standard uncertainty. Then we have the relative uncertainty given here. And this column here shows you the square root of relative uncertainty. The reason I’m giving this here is later on is I’m going to combine all the relative uncertainty by taking the sum of squares of this and making a square root of that. So in order to facilitate, plugging in of this value into my final equation I’m showing in my table all the time the square of the relative uncertainty.
Now the second component for the uN1 is making up to volume in a thousand ml flask. There are 2 uncertainties associated with this preparation. There is a tolerance associated with the flask which was given by the manufacturer as .30. As I mentioned earlier, this follows a triangular distribution so you divide this by square root of 6 and you arrive at this value. There is a temperature coefficient of expansion for glass, for the pipette that is usually given by this formula. This bar is 2.1, 10 to the -4 hard degree celsius per ml. And the variation is 3 degree and the total volume is 1000 milliliter so you plug in all these value into this equation, you would get this value. So the combined uncertainty for the flask is a combination of u tolerance or u volume plus u temperature, so these are taken from the earlier slides and you arrive at this value. Now you have to calculate the relative uncertainty. In the relative uv uncertainty, the value is thousand milliliter that’s what we used here. And this table once again puts in a nutshell all these values. The value is thousand ml, the uncertainty from the tolerance is given here, the temperature is given here, and you combine these two is given here. And the relative uncertainty is this divided by the value which is given here.
So the third component is the SRM certification. As I told you we use the SRM 351a. The certificate stated in 99.97 impurity + / - 0.014 as the uncertainty with the coverage factor 2.08. So you divide the expanded uncertainty by the coverage factor to arrive at the standard uncertainty. Now you calculate the relative uncertainty here. So this is again the table that shows you the certified value from the SRM 99.970, expanded uncertainty from the certificate value 0.014, coverage factor was 2.08, standard uncertainty calculated from this is given here. And the relative uncertainty is the standard uncertainty divided by the value and that is given here. This is the, from the SRM certification, still component of uN1.
The fourth component is the molar mass. The listed uncertainty is taken from the IUPAC table for sodium, carbon, and oxygen. The atomic mass and the uncertainty represented in the IUPAC table, for sodium, atomic weight is this, quoted uncertainty is this. IUPAC table did not state any confidence interval so we use a rectangular distribution and then the normalizing factor associated with it, so that is the value here. For carbon, the atomic weight is taken from the IUPAC table and quoted uncertainty is also given there so we normalize it and convert it to standard uncertainty. For oxygen as well, the quoted uncertainty was divided by square root of 3 to arrive at this value. Now this is the atomic mass. Now sodium carbonate consists of 2 sodium atom this is NA 203 so 2 sodium atoms so the weight calculation is 2 times the atomic weight and standard uncertainty also is 2 times the standard uncertainty that we calculated. Now carbon is 1 atom so we just replicated this here. Oxygen is 3 atoms hence the weight component, mass component from oxygen is 3 times the atomic weight and the standard uncertainty is 3 times this. So you have to sum this up and your uncertainty is the square root of sum of all this. So for the mass of sodium carbonate, which is the sum of this, we have this uncertainty given here. Now this table again presents in a nutshell the whole thing. The mass is given from here, and combined uncertainty is here and relative uncertainty is the standard uncertainty or the combined uncertainty divided by the mass which is given here. Now, we had four components of interim uncertainty for this particular process of uN1 -- coming from balance, coming from volumetric flask, coming from SRM certification, coming from molar mass and these are all the uncertainties for each of these processes. And so the total uncertainty is square root of sum of squares of this. So we arrived at the total value of uncertainty for uN1 for sodium carbonate.
Now let us proceed to calculate the second component, uV1. uV1 for sulfuric acid what we did we took 50mL of sodium carbonate to analyze sulfuric acid. So there is 1 uncertainty associated with this, represented by this symbol uV1 which is the pipette. Pipette again, I have explained this process earlier for uN1. So the listed uncertainty from the manufacturer, normalization factor is used for triangular distribution. So listed uncertainty divided by the square root of six will give you the uncertainty for u tolerance. Now it has a temperature coefficient component which again we plugged in the 50mL that we used on the same equation that we used before, so we arrived at 0.0182 as our value. Now, we have to combine both uncertainty that is u tolerance and u temperature for the pipette. So when you combined this, you arrive at this value here. So the relative uncertainty here is the combined uncertainty divided by the value which is 50mL, and combined uncertainty is the combination of u tolerance and u temperature. And relative uncertainty is combined uncertainty divided by the value which is given here.
So now we have to proceed to calculate the uncertainty of the third component, uV2. To analyze the sulfuric acid, what we did, we took the sulfuric acid in a burette and we titrated from the burette to our sodium carbonate volume that I said earlier. So there is one uncertainty associated with this symbol uV2 that is coming from the burette. So again, the principle is very similar to the calculation of the pipette uncertainty. You have a listed uncertainty for the burette from the manufacturer and it follows a triangular distribution so your u tolerance is your listed uncertainty divided by the square root of 6 and you have the temperature component which again is the standard equation that you follow. Plug in the value and this 39.2 was our burette volume so you plug in that here for the volume and you arrive at this value here which is 0.014258. Now, for the burette component, we have to combine the interim uncertainties of tolerance and temperature as we did for the pipette. So we arrive at this value. Now we have to calculate the relative uncertainty for this process and again the value here is 39.21, the u volume and the u temperature which makes up the combined uncertainty for this particular process is given here. So the relative uncertainty is the combined uncertainty divided by the value which is expressed here.
So there is one more component for here that we have to consider. When we did this normalization of sulfuric acid, we did a number of replicates. We took about 7 replicates and then we calculated the average from each of these replicates we arrived at this value. Now the standard deviation for this particular process also we calculated based on our equation of standard deviation which is given here. So the uncertainty due to repeatability, this is because of the repeatable titration, we follow the Type A. So remember the Type A that we used the standard deviation and the normalization factor as the number for 7 replicates that we used so we plug in this, the standard deviation is plugged in from the earlier equation that we had and the number of duplicates we had and we calculate the uncertainty here. The relative uncertainty is the standard uncertainty divided by the value. The value was the normality, the x bar that we arrived at from the earlier slide. So you divide that and you get the value of relative uncertainty from this here.
Now, we completed quantifying uncertainty from each of the process. What were the process, uN1 - uncertainty of sodium carbonate, uV1 - uncertainty from the volume delivered from the sodium carbonate, uV2 - uncertainty of sulfuric acid volume, repeatability of the measurement itself. So the total is, the uncertainty that you should calculate is the square root of sum of these and that is expressed here. And the normality that we found by our average was this. So this normality is multiplied by the uncertainty that we got here to arrive at the uncertainty associated with uN2 which is 2.920x10-5.
Now, only one task remains which is to expand this uncertainty. I used the standard coverage factor 2 at 95% CI and multiplied the uncertainty that we arrived at earlier and I expanded it here. So the normality of sulfuric acid that we got here is 0.02499 plus or minus 0.00006N. Expressed in terms of percentage is plus or minus 0.23%.
Now, we have various components for the interim uncertainty. We want to know which contributed significantly to our relative uncertainty. So what we did, here we charted the relative uncertainty from each task such as the flask, balance, SRM certificate, sodium carbonate, sulfuric acid, and repeatability and molar mass, we charted this. As you can see, the significant contribution is from the repeatability, volume of sulfuric acid and volume of sodium carbonate that we delivered as well as the flask. Balance and the certificate did not have much insurance. But as you can see the molar mass did not contribute much at all. It is not at all significant. This is the case in many of calculation that I have done. If you want, you could omit the including of molar mass, the uncertainty from the molar mass from your calculation but you have to justify why you removed it. So, that is the reason I am giving you this more calculation of molar mass is quite tedious, if you want you can remove it, but you may want to justify this as well.
So in summary, using the process approach and breaking it into five steps I have discussed above, you can calculate uncertainty quite easily. It’s not so difficult as it seems in the beginning. This presentation indicates general methods for estimating uncertainties in analysis. It’s by no means comprehensive. It depends on conditions and nature of analysis. We used the information given in these references to arrive at our calculation. This presentation gave focus on titration. We are planning a series of presentation focusing on methods, preparations, etc and we will be making them available to our customers. If you want any particular process to be done, please contact our marketing and sales and they would include that in our future presentation. Thank you very much for listening to this presentation and I welcome any questions now.
Thank you very much, Vanaja. We do have some time for some questions and I encourage you to ask any questions you may have by entering them on the question box on your screen but Vanaja will answer a few questions now.
The first question is, what would be your approach on qualitative methodology such as the ones found in microbiology?
So the question was, what will be your approach on qualitative methodology such as the ones found in microbiology? Uncertainty calculations are dependent on practice used on by the lab. Based on the practice, you can probably estimate whether they are type A or type B. And if they are type B, you can also figure out where it fits in the distribution norm. So if you define all these and use the the five point approach, the same approach as I have discussed in the presentation you can arrived at the calculation. If you have a particular process in mind, you may want to discuss this further with our marketing and sales.
Okay, another question that we have is how did you know what to use type B uncertainty for the flask, balance, and pipette? And how do you know when to use the rectangular versus normal versus triangular distributions.
The question is, how did you know to use type B uncertainty for the flask, balance, and pipette. How do you know when to use rectangular versus normal versus triangular distributions? Did I get this correct, Amy? Type B uncertainty for class A volumetrics would follow triangular distribution. Class A products are calibrated by the manufacturer by measuring the calibrated volume quite a number of times hence the values are likely to lie closer to the target value. You would think the distribution is greatest at the target value and falls off slowly towards the end of the range so it would follow your triangular distribution. That is why you divide it by square root of 6 resulting in the smallest standard deviation as compared to other distributions such as rectangular where you divide it by square root of 3. Usually the class A pipette the manufacturer uses here, very tight tolerance and to do that he uses a number of measurements. The next question was how to use, how to know rectangular versus normal versus triangular. You would use rectangular distributions when you have limits taken for your value without specifying the level of confidence. You have no idea where the range of the uncertainty lies. So for an instance, the two values have an equal probability of being anywhere in the range. Hence, dividing by the square root of 3 will give you the largest standard of uncertainty to counter the effect of lack of information so you would use a normal distribution.
The next question is do I have to consider the uncertainty or the purity of the water used in the preparation of sodium carbonate solution.
Let me repeat this, do I have to consider the uncertainty or the purity of the water used in the preparation of sodium carbonate solution. In a laboratory, usually, we used reagent grade water… we use wdi-less water. So here, when we use a reagent water you would expect there won’t be anything that is affecting the purity of sodium carbonate. So usually, it takes about 15 ppt of carbon dioxide.
How about what is the appropriate number of replicates needed when using the standard deviation curve at 95% confidence level. Is it a minimum of n=7 as you stated or more like n=20 is ideal?
The question is, what’s the appropriate number of replicates needed when using the standard deviation curve at 95% confidence level. Is it a minimum number 7 or number 20? There are many experiments done what is the ideal number of replicates that you have to use. Many times, there is no value added when you do replicates of more than 7. If you have an established method and you have done that so many times in the laboratory, even 3 gives you a very good idea and gives a better average. So I took 7 because it is easy to calculate the coverage factor by using student’s table. Now, if you have a well-established method and you are valid a number of times and your standard deviation is not very dispersed, I would think that you could use anything below 7 and use the student’s t for your calculation of coverage factor.
Another question we have from the audience, if this normality of sulfuric acid is used in a subsequent titration of an acid, how would you combine the uncertainties of the normality of the sulfuric acid in determination of the acid concentration to achieve an overall uncertainty associated with the acid determination.
Good question. The question asked, you used this sulfuric acid that you standardized for subsequent titrations and what would you do. Now, your sulfuric acid that you had prepared, and standardized, and quantified the uncertainty becomes almost your reference material. So you have an uncertainty associated with this and you have calculated it. Now, use this for your subsequent process. The other process that you have may involve further dilution of something, preparation of your sample that you want to titrate, consider all those, and as I have stated in my case the SRM purity that I used, the uncertainty from the SRM purity that I used substitutes the uncertainty that you arrived for your sulfuric acid in that place as a placeholder.
How can you include the uncertainty associated with different analysts in a calculation of overall uncertainty or our analyst affects the minute from uncertainty determinations.
Again, this is kind of a repeatability, you know it varies from operator to operator. It varies quite a lot. One operator doing 7 replicates is totally different from 7 operators doing the same 7 replicates. So you would find the contribution from the repeatability quite enormous. So you have to take into consideration as the repeatability uncertainty from each operator and then combine them to get the overall uncertainty for this particular process.
Is there any perfect standard distribution or uncertainty?
Again, perfect standard, each process, the uncertainty, the overall uncertainty depends on the process and depends on so many things so if you want, many times you have seen, if you use a very tightly controlled process, the uncertainty is low. And again, can I say uncertainty as low as .1%, .01% is better than uncertainty .08%? I can’t say that because it varies on the nature of the experiment. What, for the same experiment, if you conduct on tightly controlled conditions and you arrived at a better uncertainty, that’s the value that you should use. But there is no figure I can give you that this is the standard uncertainty or you should maintain within that. As I told you earlier in the presentation, uncertainty is not, if you have more uncertainty doesn’t mean that you have a problem. It gives you only confidence in your measurement. It doesn’t give you any doubt. This is what I said in the beginning of my presentation.
When making our estimations of uncertainty, is it okay to approximate or assume some uncertainties are low and therefore negligible in our estimation of overall uncertainty.
Yes. In the last slide that I presented to you, I had put a chart showing the contribution from the various tasks and then I have shown you the contribution from the molar mass was very insignificant compared to all of the other uncertainties. So if you want it, you can, if you want it that, in your future calculations I said that what happens is, sometimes, your regulating body may say, did you consider it. So you may want to have this done once or twice and then show your regulator or auditor or whoever, “Okay I have done this. It does not giving me significant contribution so I am going to omit it in my future calculations.” And to my view that is a valid argument.
Okay, I think we’re going have one more question. In your example, why did you use triangular distribution to estimate uncertainty from volume measurements but a uniform distribution to estimate uncertainty for mass measurements.
Uhm, the question was why did I use triangular distribution for volume measurements and uniform distribution for mass measurements. The triangular distribution, I used a class A pipette. Class A pipette comes with very well specified tolerance. So I know that uncertainty component from that is quite low. So if I use the square root of 6 as my uncertainty normalizing factor, I will arrive at a very low uncertainty due to the glassware. However, in the mass I have said in the IUPAC table that I used the atomic weights and the uncertainty associated with the atomic weights. IUPAC table did not tell me what confidence interval that was so I have no choice but to use a rectangular distribution which gives a greater uncertainty value to account for the lack of information about the confidence limit.
Okay, I think that’s about all time that we have today. Thank you very much for the questions you sent for us today. I’d like to thank you all for attending. Just a quick reminder that we’ll be sending everyone a copy of the presentation. We’ll also be sending an email with the link to the recording of the webinar. And we very much appreciate your time and we look forward to seeing you back at future webinars. Thank you and have a good day!